
product

科腾 全自动凝胶色谱系统GPC-200产品特点:1.全返控的色谱工作站软件,即除数据采集处理外,仪器各部件的控制参数均可在软件的界面中进行…
详情>>

科腾 在线天然气分析仪 BGC-4500简介产品特点:1. 24小时在线监测,实时上传数据。2. 采用西门子PLC控制,RS485和TCP/IP通信。3. 自动生…
详情>>

科腾 激光粒度仪 KTW-1000简介KTW-1000采用先进的散射原理和会聚光傅立叶变换光路。高密度探测器及全量程无缝衔接测试方法,保证了测试结…
详情>>

科腾 化学吸附仪 JK-H2000简介主要功能:TPD、TPO、TPR、TPS、脉冲滴定、储氧量分析、比表面积测定1.两位六通阀,用于脉冲滴定法分析金属…
详情>>

科腾 比表面积分析仪 KT-4000产品特点:比表面积分析仪 KT-4000 应用领域能源与材料:电池材料用于测量锂离子电池正负极材料(如石墨…
详情>>

科腾 比表面积分析仪 KT-3000产品特点:2.智能选择多容量空间技术。3.全不锈钢真空系统连接密封采用金属密封技术。4.真空系统内壁采用镀…
详情>>

科腾25位热解析ATD-01简介产品特点:25位热解析 ATD-01 应用领域25 位热解析仪的应用领域非常广泛,主要包括以下几个方面环境检测、材料…
详情>>

科腾1位热解析JK-0A简介产品特点:1 位全热脱附-解吸仪气路采用电动阀和电磁阀相结合,采用触摸屏方便编辑、保存、调用吸附管的解吸、进…
详情>>

科腾20位顶空进样器JK-02简介产品特点:1、开机自检,故障报警和提示,样品盘自动定位,屏幕上可以实时显示当前运行样品号、起始样品号。…
详情>>
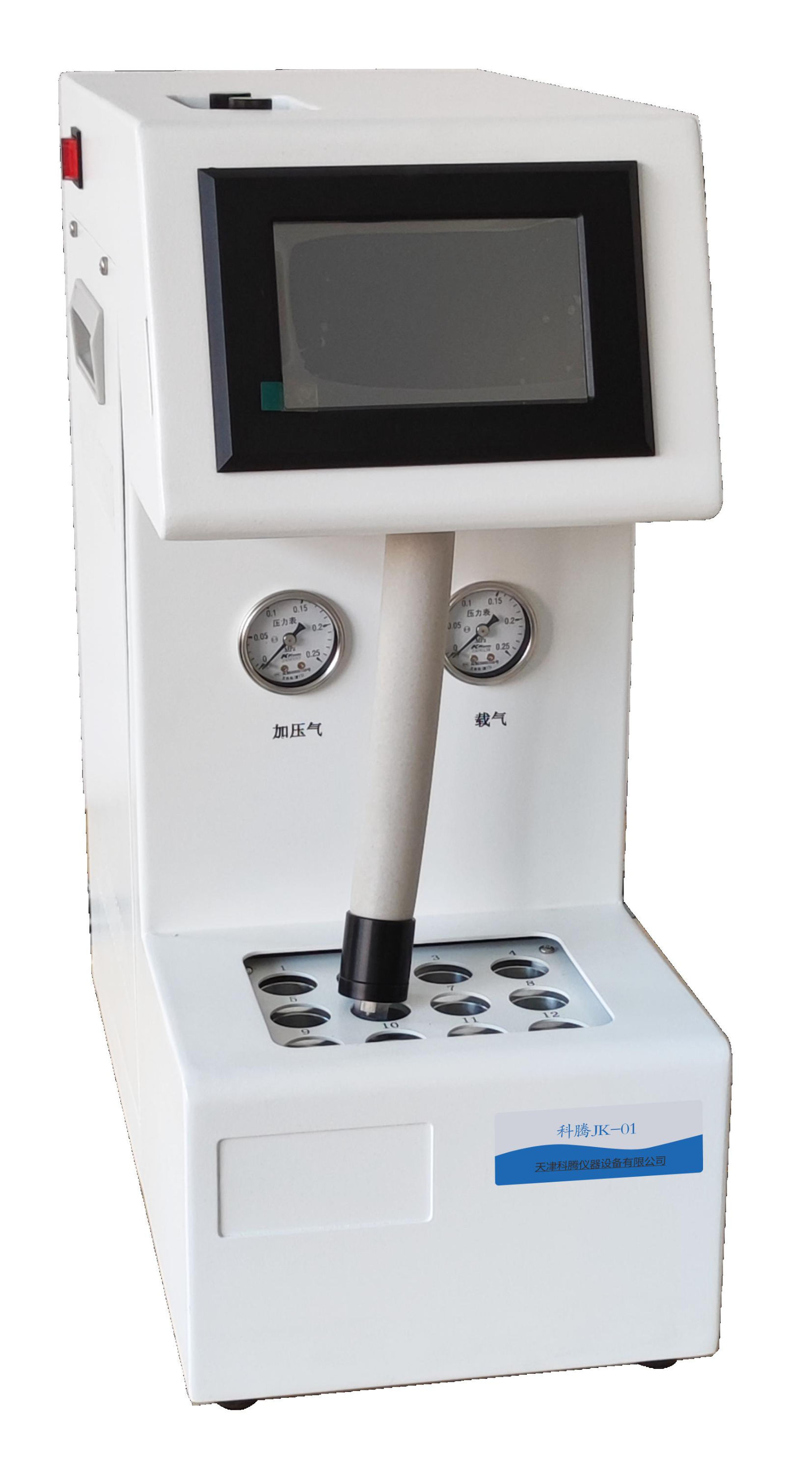
科腾12位顶空进样器JK-01简介产品特点:▲5寸彩色触屏,界面友好,清晰简洁,无需花太多精力即可上手,方便使用者快速操作;▲人性化结构…
详情>>

科腾纯水机JUP20L简介主要性能特点:1.全塑外壳,模具一次成型2.微电脑控制 LED 彩屏显示,全自动产水3.双出水,可同时出 UP 水和 RO 水…
详情>>

科腾纯水机JUP10L简介主要性能特点:1.全塑外壳,模具一次成型2.微电脑控制 LED 彩屏显示,全自动产水3.双出水,可同时出 UP 水和 RO 水…
详情>>
关于开展“中国机械工业联合会机械工业人才评价中-心仪器仪表实训基地”职业能力评价培训课程通知一、培训内容1、 仪器培训类别:傅里叶红外光谱、ICP等离子体发射光谱。2、 仪器培训内容:仪器理论培训、使用操作及简单维护培训、实操培训。3、 “分析仪器…

关于开展“中国机械工业联合会机械工业人才评价中-心仪器仪表实训基地”职业能力评价培训课程通知一、培训内容1、 仪器培训类别:傅里叶红外光谱、ICP等离子体发射光谱。2、 仪器培训内容:仪器理论培训、使用操作及简单维护培训、实操培训。3、 “分析仪器…
北京京科瑞达主营二手分析仪器、仪器租赁、维修维保服务
Copyright © 北京京科瑞达科技有限公司 版权所有 工信部ICP备案号:京ICP备09012202号-5
服务热线













